Patogeneza infekcji Helicobacter pylori i wybrane metody leczenia
WSTĘP
Helicobacter pylori jest urzęsioną gram-ujemną pałeczką należącą do rzędu Campylobacteriales. Została po raz pierwszy wyizolowana w 1899 roku przez Walerego Jaworskiego, ale dopiero w 1983 roku Barry Marshall i Robin Warren udowodnili jej patogenność przez wypicie zawiesiny bakterii co spowodowało ostre zapalenie żołądka. Do swojego wzrostu wymaga obecności dwutlenku węgla przez co nie jest łatwa do hodowli in vitro.
Patogen bytuje w układzie pokarmowym człowieka i zwierząt domowych, a kolonizuje przede wszystkim powierzchnię komórek epitelium trzonu i części około-odźwiernikowej żołądka. Ponadto, można ją spotkać również w ślinie, płytce nazębnej, w dwunastnicy, żółci i w kale. Natomiast do infekcji dochodzi głównie od innych ludzi lub zwierząt domowych, ale źródłem zakażenia może być również nieprzegotowana woda. Dzieje się tak, ponieważ kokoidalna forma bakterii ma zdolność do tworzenia biofilmu na powierzchni kranu i rur wodociągowych. Również spożywanie niemytych warzyw i owoców oraz surowego mięsa może stać się źródłem zakażenia.1,2
Epidemiologia
Światowa Organizacja Zdrowia szacuje, że 20-50% populacji krajów rozwiniętych i aż 80% populacji krajów rozwijających jest zakażona H. pylori. Natomiast związane jest to przede wszystkim ze złymi warunkami sanitarnymi. Polskie Towarzystwo Gastroenterologiczne podaje, że bakteria obecna jest u 70% społeczeństwa naszego kraju.1
FORMY WYSTĘPOWANIA
Wyróżnić można trzy formy występowania Helicobacter pylori:
- spiralna – forma zaraźliwa, wirulentna i zdolna do wzrostu w warunkach laboratoryjnych. Jest wykrywana u pacjentów z wrzodami dwunastnicy lub zapaleniem żołądka.
- kokoidalna – forma mniej wirulentna i nie hodowana w warunkach laboratoryjnych. Powstaje z formy spiralnej w niesprzyjającym środowisku i posiada podobny zestaw białek błonowych i cytoplazmatycznych.
- zdegenerowana kokoidalna – posiada częściowo zdegradowany materiał genetyczny, osłabioną strukturę błony komórkowej i zdegenerowane adhezyny i poryny.1,2,4
TWORZENIE BIOFILMU
Biofilm – to trójwymiarowa heterogenna struktura złożona z drobnoustrojów zawieszonych w ich wytworach macierzy zewnątrzkomórkowej, mających zdolność adhezji do powierzchni stałych i do siebie nawzajem. Tworzona może być przez wiele patogenów i umożliwia przetrwanie w niesprzyjających warunkach oraz na wymknięcie się z odpowiedzi układu odpornościowego człowieka. Ponadto, biofilm tworzony przez Helicobacter pylori zawiera egzopolisacharydy umożliwiające kolonizację i odporność na zmiany pH oraz nadające strukturze biofilmu wytrzymałość. W dodatku, biofilm tworzy barierę znacznie osłabiającą działanie antybiotyków.
Podobnie jak u innych mikroorganizmów tworzących biofilm, zachodzi tu zjawisko quorum sensing (rodzaj komunikacji) za sprawą wydzielania autoinduktorów (rola genu luxS i cagE). Zachodzi wymiana informacji genetycznej na plazmidach, przez co bakterie współdzielą czynniki wirulencji. Następować może przez to zmiana struktury biofilmu w raz z gęstością komórek (np. w odpowiedzi na dostępność składników pokarmowych).2
OBJAWY CHOROBOWE I CHARAKTERYSTYKA INFEKCJI
Głównym objawem zakażenia H. pylori jest zapalenie błony śluzowej żołądka. Przy czym, wyróżnić można: fenotyp łagodnego zapalenia błony śluzowej żołądka, fenotyp wrzodowy i fenotyp nowotworowy. Zmiany zapalne mogą prowadzić do atrofii i metaplazji błony śluzowej żołądka czego konsekwencją może być choroba wrzodowa lub nowotwór komórek epitelium żołądka (również chłoniak MALT – nowotwór tkanki limfatycznej związanej z błoną śluzową układu pokarmowego). Jednakże, częściej obserwuje się fenotyp nowotworowy jako konsekwencję zanikowego zapalenia żołądka. Obserwuje się również chorobę wrzodową dwunastnicy, jednakże w takim przypadku nigdy nie dochodzi do powstania nowotworu żołądka.
Przebieg infekcji
Bakteria po wtargnięciu do światła żołądka i przekroczeniu błony śluzowej, dociera do komórek nabłonka żołądka dzięki całej puli toksyn, enzymów i składników ściany komórkowej oraz wytworów macierzy zewnątrzkomórkowej. Następnie, po adhezji do komórek tworzyć może strukturę biofilmu, gdzie jest odporna na działanie układu odpornościowego i leków. Może przetrwać również wewnątrz komórek odpornościowych (wewnątrz fagosomu).
Z antrum żołądka, po przedostaniu się przez odźwiernik do dwunastnicy, patogen może dalej migrować i zostać wydalony z organizmu lub zasiedlić dwunastnicę przyczyniając się do powstania stanu zapalnego. Może tutaj przyczynić się do zaniku błony śluzowej (fenotyp wrzodowy). W przypadku wymiotów może pozostać w jamie ustnej w formie kokoidalnej, wewnątrz płytki nazębnej, co prowadzić może do wtórnego zakażenia.

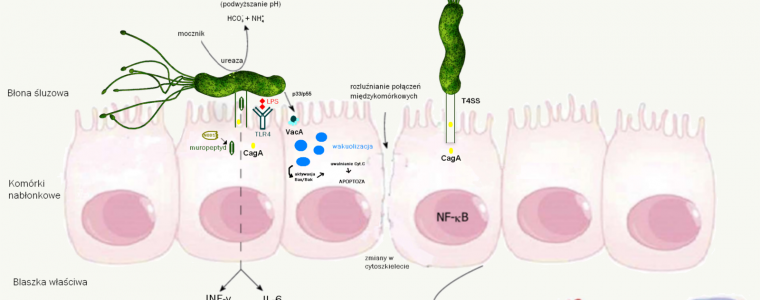
Rysunek 1. Infekcja Helicobacter pylori i odpowiedź immunologiczna w komórkach nabłonka żołądka. CagA: białko A związane z cytotoksyną; VacA: cytotoksyna wakuolizująca; LPS – lipopolisacharyd; TLR4: receptor Toll-podobny 4; NF-κB: czynnik jądrowy-kappaB; IL: interleukina; INF-γ: interferon-γ; TNF-α: czynnik martwicy nowotworu-α; NOD1: białko 1 zawierające domenę oligomeryzacji wiążącą nukleotyd.
GŁÓWNE CZYNNIKI WIRULENCJI
Do czynników wirulencji produkowanych przez Helicobacter pylori zaliczyć można m.in. białka umożliwiające kolonizację, neutralizację kwaśnego pH, adhezję do komórek żołądka. Ponadto, czynniki umożliwiające modyfikacje metabolizmu epitelium, inicjację stanu zapalnego w komórkach oraz ucieczkę od odpowiedzi immunologicznego.1,2
- Ureaza – enzym przeprowadzający rozkład mocznika do CO2 i amoniaku, który powoduje neutralizację działania kwasu solnego w żołądku
- Mucynaza – indukuje degradację struktury mucyny (składnika błony śluzowej). Ułatwia to bakterii przekroczenie ochronnej warstwy błony śluzowej i inwazję komórek epitelium żołądka
- LPS – Lipopolisacharyd – składnik ściany komórkowej, który stymuluje układ odpornościowy do wytwarzania cytokin prozapalnych, co w konsekwencji zaburza homeostazę niszy błony śluzowej żołądka m.in. poprzez zahamowanie syntezy i wydzielania mucyny.
- Rzęski – umożliwiają ruch i przemieszczenie się bakterii do warstwy podśluzowej (występuje od 2 do 7 rzęsek).
- Enzymy rozkładające reaktywne formy tlenu: Katalaza, dysmutaza ponadtlenkowa – niwelują atak komórek odpornościowych.
- Cząsteczka adhezyjna Bab A – wiąże się z antygenem Lewis b na epitelium34
- HP-NAP (H. pylori Neutrophil Activation Protein) – białko aktywujące neutrofile
- OipA (Outer inflammatory protein A) – zewnętrzne białko zapalne A
- DupA (Duodenal ulcer promoting A) – białko odpowiedzialne za powstawanie wrzodów dwunastnicy
- T4SS (Type Four Secretion System) – system sekrecji typu IV – molekularna “strzykawka” odpowiadająca za przekazywanie białek efektorowych z cytozolu Helicobater pylori do wnętrza zakażonych komórek, w tym m.in. białek CagA i produktów rozpadu peptydoglikanu (muropeptydy). W adhezji T4SS do komórek gospodarza uczestniczą m.in. obecne na końcu pili białka CagL i CagY, które łączą się z integrynami (α1β1 – receptora dla kolagenu i lamininy i α5β1 – receptora dla fibronektyny) na powierzchni komórek nabłonkowych. Poprzez transport fragmentów peptydoglikanu system sekrecji typu IV przyczynia się do aktywacji wydzielania prozapalnej interleukiny 8, wytwarzania metaloproteaz i genów antyapoptotycznych (poprzez przyłączenie się do receptora NOD-1 i aktywację NF-κB).
- CagA (Cytotoxin associated protein A, cytotoxin-associated gene antigen) – białko o masie 120-145 kDa, produkowane przez ok. 60% izolatów H. pylori. Razem z cytotoksyną wakuolizującą (Vac A) pełni kluczową rolę w patogenezie zakażenia (m.in. ostre lub podostre zapalenie żołądka, zanikowe zapalenie błony śluzowej żołądka i gluczolakorak żołądka). Wprowadzana jest do cytozolu organizmu poprzez kompleks białkowy – T4SS (rola interakcji z integryną obecną na powierzchni epitelium żołądka). Oddziałuje tam z białkami gospodarza wpływając na szlaki sygnałowe zależne i niezależne od fosforylacji. Szlaki te regulują m.in. proliferację, ruchliwość i kształt komórek (poprzez zmiany w cytoszkielecie i osłabienie ścisłych połączeń międzykomórkowych). Powodują również, transkrypcję wielu genów, w tym powodujących ekspresję cytokin prozapalnych (IL-6, IL-8, Interferon-γ, TNF-α). Ponadto, białko CagA jest protoonkogenem (zwłaszcza jej forma zawierająca na C-końcu motyw EPIYA-A-B-D), z uwagi na wpływ na aktywność kinaz regulujących przemiany cytoszkieletu, proliferację i różnicowanie się komórek (wiele genów tych kinaz należy do onkogenów)
- VacA – wakuolizująca cytotoksyna, obecna u wszystkich szczepów. Protoksyna o masie 140 kDa kilkakrotnie ulega cięciu z utworzeniem dwóch głównych podjednostek, z których p33 odpowiada za wbudowanie się do błony komórkowej, a p55 za związanie się z nią. Następnie, powstaje kanał jonowy, który po fuzji z endosomem i przyłączeniu się do błony wakuoli, może utworzyć w niej kanał jonowy zwiększający przepuszczalność dla niektórych kationów (np. Fe3+ i Ni2+) i niewielkich molekuł organicznych. Przyczynia się to do napływu wody i powstawania większej ilości wakuol (wakuolizacja). jednocześnie VacA powoduje pęcznienie komórki, ale także inicjuje zmiany w cytoszkielecie, rozluźnia ścisłe połączenia międzykomórkowe, hamuje proliferację komórek epitelium żołądka oraz może zaburzać mitochondrialny potencjał transbłonowy, doprowadzając w ten sposób do uwolnienia cytochromu C do cytoplazmy, w wyniku czego dochodzić może do apoptozy, jednakże proces ten jest przeważnie hamowany przez białko CagA.8,9 Toksyna VacA ma również swój udział w zaburzaniu odpowiedzi immunologicznej, ponieważ zakłóca fagocytozę, hamuje proliferację i skuteczność działania limfocytów T oraz prezentację antygenu.
ONKOGENEZA
Helicobacter pylori odpowiada za powstanie dwóch typów nowotworów żołądka: chłoniak (mięsak) i gruczolakorak (adenocarcinoma), przy czym nigdy nie występują one jednocześnie.
Chłoniak typu MALT
Początkowy pseudochłoniak (pseudolymphoma) czyli typ nisko-złośliwego nowotworu rzekomego, powstałego z udziałem limfocytów poliklonalnych (pojawia się punktowy naciek limfocytów), może ulec przeobrażeniu do chłoniaka typu MALT (MALToma – nowotwór typu mezenchymalnego), który jest nowotworem jednego typu limfocytów – najczęściej limfocytów B. W takim przypadku dochodzi do niekontrolowanych podziałów w wyniku zmian w materiale genetycznym (translokacja, trisomia i mutacja m.in. genów uczestniczących w cyklu komórkowym, np. c-myc). Natomiast forma wysokozłośliwa pojawia się m.in. po delecji genu TP53 – kodującego czynnik transkrypcyjny p53, który wykazuje właściwości supresora nowotworowego (jest to regulator cyklu komórkowego).
Nowotwór komórek epitelium żołądka
W mniejszym stopniu H. pylori odpowiada za indukcję nowotworu komórek epitelium żołądka (nabłonkowca), jednakże jest jego najczęstszą przyczyną (90-95% przypadków nowotworu nabłonkowego żołądka, oprócz wirusa Epsteina-Barr’a). Powstaje on głównie na skutek stymulowania przez patogen wydzielania cytokin, czynników wzrostu i reaktywnych form tlenu. Dochodzi do zaburzenia równowagi między procesami powstawania nowych komórek a ich apoptozą, np. podczas zanikowego zapalenia żołądka, zaniku śluzówki i zmian w fenotypie: metaplazja jelitowa (komórki nabłonka żołądka zamieniają się w komórki nabłonka jelit) lub dysplazja. Tutaj również dochodzi do niekontrolowanych podziałów w wyniku wyłączenia genu TP53, ale ponadto zaobserwowano nadaktywację genów: c-met (po translacji powstaje receptor czynnika wzrostu hepatocytów), c-erbB2 i k-sam (w wyniku ich ekspresji powstają białka o aktywności kinazy tyrozynowej) i mutację w genie kodującym E-kadherynę.34
Udział białka CagA
W procesie onkogenezy największe znaczenie ma białko CagA. Uznawane jest przez Międzynarodową Agencję Badań nad Rakiem (ang. IARC – International Agency Research on Cancer) za karcynogen grupy I. W komórce epitelium tyrozyna motywu EPIYA zostaje ufosforylowana przez kinazy SFKs po czym oddziałuje z białkiem zawierającym domenę fosfatazy tyrozynowej SHP-2. Interakcja ta powoduje nadaktywność fosfatazy SHP-2, a w konsekwencji również zwiększoną aktywację kinaz Erk/MAP i zwiększenie nieufosforylowanej czyli nieaktywnej kinazy FAK (regulatora kształtu i ruchliwości komórki), co przyczynia się do zmian fenotypu komórek nabłonka żołądka: rozregulowania cyklu komórkowego, wydłużenia kształtu i rozproszony wzrost komórek oraz modulację odpowiedzi immunologicznej.
Toksyna CagA po fosforylacji oddziałuje również z białkiem Crk, które w formie aktywnej również aktywuje kinazy Erk/MAP, co dodatkowo zaburza cykl komórkowy przyczyniając się do powstawania zmian nowotworowych m.in. poprzez aktywację czynników tranksypcyjnych promujących proliferację (w tym ELK1 – E-26 like protein 1). Ponadto, oddziaływanie CagA z ludzkim białkiem Crk powoduje reorganizację cytoszkieletu aktynowego (udział białka Rac1 – ras-related C3 botulinum toxin substrate 1) oraz rozluźnienie połączeń międzykomórkowych (udział Rac1 i WAVE – WASP family Verprolin-homologous protein).1
Obecność aktywnej formy białka CagA prowadzi do apoptozy komórek epitelium żołądka i ubytków w błonie śluzowej, co ze względów bytowania H. pylori jest niekorzystne. Dlatego też, bakteryjne białko CagA musi posiadać enzym regulujący jego aktywność – jest nim kinaza Csk, która inaktywuje kinazy Src aktywujące CagA, co opóźnia zmiany morfologiczne i apoptozę zainfekowanych komórek nabłonka. Innym mechanizmem regulacyjnym jest zmiana konformacyjna białka Src po uforsforylowaniu białka CagA czyli zmniejszenie puli aktywnej formy kinazy Src. Skutkiem ubocznym jest tutaj obniżenie ilości aktywnej formy białka biorącego udział w polimeryzacji i przebudowie cytoszkieletu aktynowego – kontaktyny (substratu kinazy Src).1
Białko CagA oddziałuje z cząsteczkami efektorowymi człowieka również na drodze fosforylacyjno-niezależnej. Przykładem jest tu interakcja z PAR1b (ang. Partitioning-defective 1b, syn. MARK2 – Microtubule Affinity-Regulating Kinase2) – kinazy serynowo-treoninowej, która warunkuje powstawanie połączeń zwierających pomiędzy komórkami jednowarstowego nabłonka. Dzieje się tak ponieważ, CagA powoduje przetransportowanie białka PAR1b z części lateralnej do części szczytowej błony komórkowej co powoduje zaburzenie połączeń międzykomórkowych, polarności komórek, zmiany fenotypu komórek i zahamowanie powstawania mikrotubul. Warto również wspomnieć, że blokada oddziaływania CagA z PAR1b powoduje jego kierowanie na drogę degradacji, co można wykorzystać w terapii nowotworu żołądka.
Innym przykładem jest aktywacja NFAT (ang. nuclear factor of acitvated T-cells) poprzez oddziaływanie CagA z białkiem Grb2 (ang. growth factor receptor-bound protein 2) w wyniku czego dochodzi do aktywacji szlaku kinaz Ras/MAP (VacA również może aktywować NFAT). Ponadto, skutkiem aktywacji NFAT jest spowolnienie podziałów komórkowych i zmiana w ruchliwości komórek.1
Innym białkiem, którego oddziaływanie z CagA zwiększa prawdopodobieństwo powstawania zmian nowotworowych jest β-kantenina. W normalnych warunkach występuje ona w błonie w kompleksie z E-kadheryną, biorąc udział w tworzeniu połączeń zwierających. Białko CagA uwalnia β-kanteninę z połączeń z E-kadheryną, co powoduje jej translokację do jądra komórkowego i aktywację genów dla czynników transkrypcyjnych biorących udział w ekspresji genów odpowiedzialnych za różnicowanie się komórek i za proliferację (zwiększenie poziomu aktywnej formy czynnika Lef/TCF). W konsekwencji, może dojść metaplazji komórek nabłonkowych i do zmian nowotworowych. Natomiast bezpośrednim działaniem proonkogenne białka CagA jest oddziaływanie z białkiem onkosupresyjnym – RUNX3 i jego degradację na skutek poliubikwitynacji.1